| Autor |
Nachricht |
Marga90
Gast
|
 Verfasst am: 20. Jan 2015 17:42 Titel: Bioinformatik: Sequenzalignments für nicht Bioinformatiker Verfasst am: 20. Jan 2015 17:42 Titel: Bioinformatik: Sequenzalignments für nicht Bioinformatiker |
 |
|
Hallo liebe Bio-Boardler,
ich studiere Mathematik und kenne mich mit Biologie entsprechend nur auf Schullevel aus.
Nun muss ich mich aber mit einem biologischen Thema beschäftigen. Zwar auf mathematischer Ebene, doch Grundlegendes muss ich ja doch erstmal verstehen.
Also Folgendes:
Ich habe 10 multiple Sequenzalignments vom Hepatitis C-Virus und nun soll jedem möglichen Paar der 2955 Aminosäure-Positionen je Alignment ein Kovarianz-Score zugeordnet werden. Ja, es handelt sich laut Paper um Aminosäuren, nicht um Nukleotide.
Der Score berechnet sich als "Quadrat der Differenz von Anzahl beobachteter und erwarteter Aminosärepaare, geteilt durch Anzahl der Einträge (außer Lücken) in jeder Spalte."
(observed minus expected squared [OMES] method, falls euch das was sagt):
^2}{N_{valid}}) mit mit
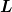 ...Liste aller beobachteten Paare ...Liste aller beobachteten Paare
 ...Häufigkeit, dass ein Paar auftritt ...Häufigkeit, dass ein Paar auftritt
und  mit mit
 ...Häufigkeit von Residue x an Position i ...Häufigkeit von Residue x an Position i
 ...Häufigkeit Residue y an Position j ...Häufigkeit Residue y an Position j
Mein Problem ist, dass ich das nicht verstehe und auch keine Vorstellung davon habe, mangels biologischer Ausbildung... Zu dem Thema Alignment hab ich mich schon ein wenig informiert und weiß, dass damit Sequenzen auf Ähnlichkeit untersucht werden. Diese Formel für den Score verstehe ich allerdings nicht.
- Was ist gemeint mit "Anzahl erwarteter Aminosäurepaare"?
- Wieso gibt es da "Lücken"?
Ich verstehe doch richtig, dass ich jedem Positionspaar einen Wert (Kovarianzscore) zuordne? Aber sind das nicht verschiedene Scores, je nach dem, welche Aminosäuren ich zugrunde lege?
Aber vielleicht verstehe ich auch etwas falsch... über eine Hilfestellung wäre ich sehr dankbar!
Viele Grüße! |
|
 |
Firelion

Anmeldungsdatum: 27.08.2009
Beiträge: 1878
|
| Verfasst am: 20. Jan 2015 19:23 Titel: |
|
|
Hi,
die Lücken entstehen durch bestimmte Mutationen die InDels also das Hinzukommen oder das Wegfallen einer Aminosäure. Wenn man animmt das Lücken vorhanden sein köönen, trägt man der Evoultion Rechnung.
Die OMES Methode kenne ich nicht.
LG Firelion
_________________
It is well known that a vital ingredient of success is not knowing that what youre attempting cant be done - Terry Pratchett |
|
|
Marga90
Gast
|
| Verfasst am: 20. Jan 2015 21:46 Titel: |
|
|
Danke für die Antwort!
Sonst noch jemand, der mir diese Score-Berechnung näher bringen kann? |
|
|
Hedera

Anmeldungsdatum: 08.03.2011
Beiträge: 657
|
| Verfasst am: 21. Jan 2015 09:58 Titel: |
|
|
Wie Firelion schon erwähnt hat kommt diese Lücken durch die annahme von Mutationen zu stande.
Im Grunde geht ein Algorithmus für Alignments so vor, dass er die beste Übereinstimmung sucht.
Nehmen wir mal an, dass bei zwei Aminosäuresequenzen eine 100 und die andere 99 Aminosäuren besitzt. Beide Sequenzen sind identisch, jedoch hat die kürzere eine weniger. Sagen wir mal position 50.
Nun werden die ersten 49 Aminosäuren übereinander gelegt. Dann folgt eine Lück bei der kurzen, gefolgt von den restlichen 50 Aminosäuren.
Das ist jetzt ein extrem einfaches Beispielt. Da man in der Regel doch recht unterschiedliche Sequenzen vergleicht kommen dann viele Lücken zustanden, weil man immer versuch die beste Übereinstimmung zu finden. Dazu kommt bei Aminosäuresequenzen, dass man ja mehr als nur 4 Möglichkeiten pro Position hat. Gibt eben mehr Aminosäuren. Und da verschiedene Aminosäuren dennoch ähnliche "Funktionen" haben, kann es auch passieren, dass verschiedene Aminosäuren auf einer Position landen.
Das ganze ist garnicht so einfach, weil man tatsächlich viel beachten muss.
Vielleicht hilft dir das hier noch weiter:
http://de.wikipedia.org/wiki/Smith-Waterman-Algorithmus
Grundsätzlich kann man den Algorithmus auch für Aminosäuren verwenden.
Und hier nochmal ein Eintrag zu multiple alignments:
http://de.wikipedia.org/wiki/Sequenzalignment |
|
|
Marga90
Gast
|
| Verfasst am: 21. Jan 2015 12:54 Titel: |
|
|
Hallo,
vielen Dank, dass ihr euch die Zeit nehmt, zu antworten.
Allerdings ist mir das Prinzip der Alignments klar. Mir geht es konkret um die Berechnung von S, dem Kovarianzscore.
Wenn der nämlich so berechnet wird, wie ich mir das vorstelle, dann ist er 0, wenn zwei Positionen in jedem Alignment gleich sind. Eigentlich müsste er doch dann maximal sein?!
Viele Grüße |
|
|
Hedera
Anmeldungsdatum: 08.03.2011
Beiträge: 657
|
| Verfasst am: 21. Jan 2015 13:47 Titel: |
|
|
Wir können gerne versuchen das hier zu lösen, aber ohne Garantie. Bei mit scheitert es eher an der Mathematik 
Wie heißt das Paper? Dann gucke ich mal was ich für dich tun kann |
|
|
Marga90
Gast
|
| Verfasst am: 21. Jan 2015 14:03 Titel: |
|
|
Alles klar 
Das Paper heißt "Genome-wide hepatitis C virus amino acid covariance networks can predict response to antiviral therapy in humans" von Aurora et al. |
|
|
Firelion
Anmeldungsdatum: 27.08.2009
Beiträge: 1878
|
| Verfasst am: 21. Jan 2015 18:31 Titel: |
|
|
Ich glaube, dass das darum geht wie wichtig es ist das bestimmte Aminosäuren nebeneinander stehen. Du zählst für jede Position wie oft welche Aminosäure dort steht. Und dann zählst du wie viele Sequenzen deines Alignments dort überhaupt eine Aminosäuren haben. Dann kannst du mit der Formel berechnen wieoft du ein bestimmtes Aminosäurepaar erwarten würdest. Dann zählst du wie oft dieses Paar tatsächlich vorkommt.
Wenn dieses Paar jetzt öfter oder seltener vorkommt als du allein durch die Häufigkeiten der jeweiligen Aminosäuren an der jeweiligen Positionen erwarten würdest, könnte das eine biologische Bedeutung haben.
Wenn z.B. die Hälfte der Sequenzen an erste Stelle ein P und die Hälfte der Sequenzen an erster Stelle ein M haben je die Hälfte der Sequenzen an zweiter stelle ein V bzw ein L haben würdest du bei 20 Sequenzen je ((10* 10))/ 20 = 5 Alignments mit PV, PL, MV und ML erwarten, oder?
Wenn du jetzt aber 7* PV, 3* PL, 7*ML und 3*MV zählst, weicht das ja von deiner Erwartung ab.
Deswegen berechnest du dann die die Differenzen und quadrierst diese.
Das wäre dann ja jeweils 4. Im Bespiel hasben wir auch 4 Paare.
Der Score wäre deswegen 16/ 20 = 0.8.
Würden wir allerdings 5 mal jedes Paar finden wäre die Differenz 0 und damit der Score auch 0.
Der Score zeigt also an wier oft es ,, fixe" Päärchen an einer Stelle gibt. Im ersten Beispiel ist es scheibbar wichtiger welche Aminosäuren ,,Nachbarn" sind als im zweiten Beispiel , wo es ,,freie" Kombination gibt.
_________________
It is well known that a vital ingredient of success is not knowing that what youre attempting cant be done - Terry Pratchett |
|
|
Hedera
Anmeldungsdatum: 08.03.2011
Beiträge: 657
|
| Verfasst am: 21. Jan 2015 20:31 Titel: |
|
|
|
Sehe ich auch so |
|
|
Firelion
Anmeldungsdatum: 27.08.2009
Beiträge: 1878
|
| Verfasst am: 22. Jan 2015 00:38 Titel: |
|
|
Das beruhigt mich  Ich war nur erst etwas wegen der Spalten verunsichert. Ich bin es gewohnt nach Matches/Missmstches zu suchen und dem entsprechend zu scoren. Ich war nur erst etwas wegen der Spalten verunsichert. Ich bin es gewohnt nach Matches/Missmstches zu suchen und dem entsprechend zu scoren.
_________________
It is well known that a vital ingredient of success is not knowing that what youre attempting cant be done - Terry Pratchett |
|
|
|


